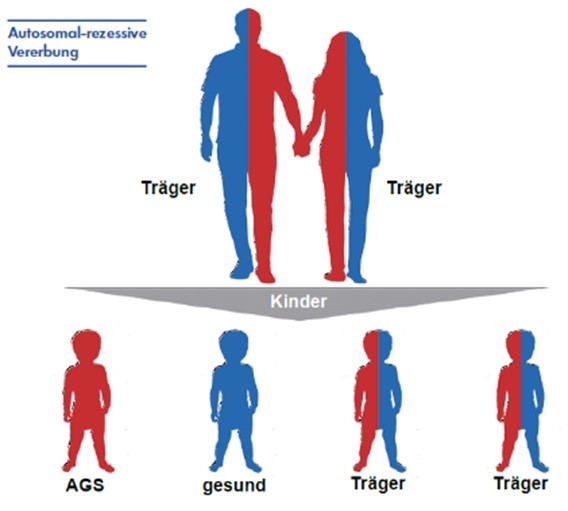
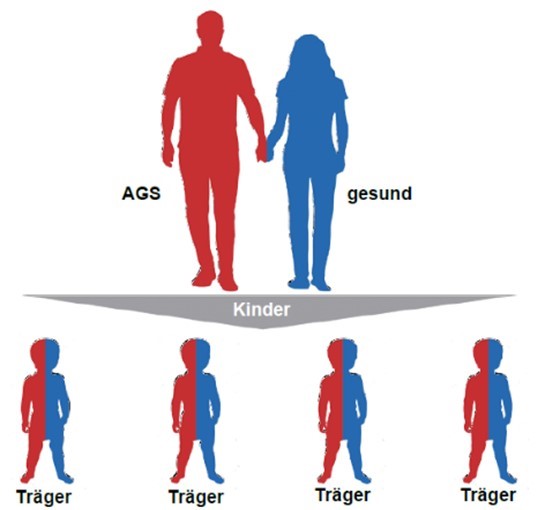
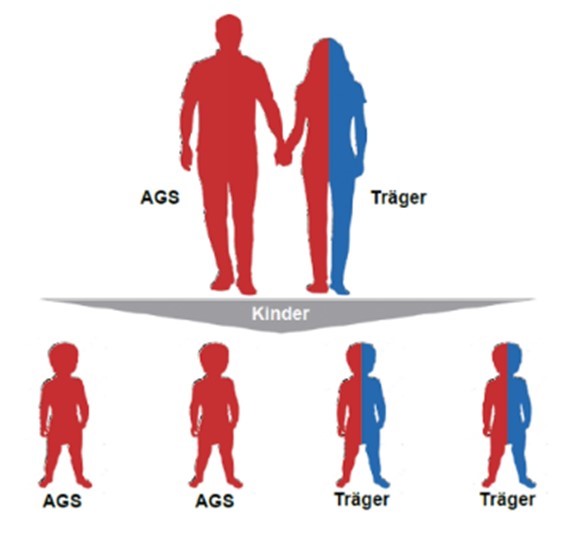
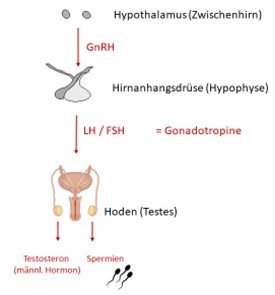
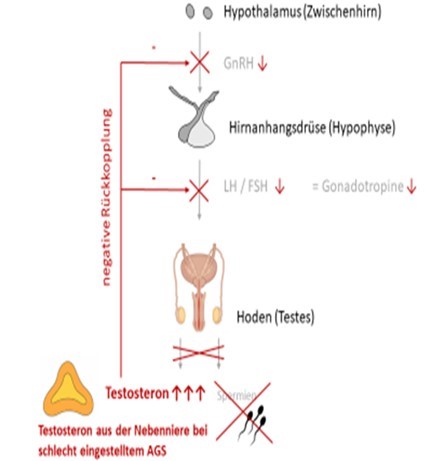
AGS bei (jungen) Erwachsenen
In diesem Abschnitt lernen sie Änderungsmöglichkeiten im Hinblick auf die Medikamentengabe ab dem jungen Erwachsenenalter sowie alle essenziellen Vorsorgeuntersuchungen kennen. Die sogenannte „Transitionsmedizin“ ermöglicht einen gelungenen Übergang von der Kinderendokrinologie zur Erwachsenenendokrinologie.
Nach Abschluss des Wachstums ist es prinzipiell möglich, von Hydrocortison auf länger wirksame, synthetisch hergestellte Glukokortikoid-Präparate wie Prednison oder Dexamethason umzustellen. Dies hat den Vorteil, dass aufgrund der längeren Wirksamkeit Prednison nur zweimal täglich und Dexamethason nur einmal täglich eingenommen werden muss. Allerdings wirken Prednison und Dexamethason deutlich stärker, sodass häufiger Nebenwirkungen wie hoher Blutdruck, Gewichtzunahme, Hautstreifenbildung (Striae), Akne und auch eine schlechtere Glukoseverträglichkeit (diabetische Stoffwechsellage) auftreten können. Prinzipiell ist das Hydrocortison dem körpereigenen Kortisol am ähnlichsten und damit auch am verträglichsten. Mittlerweile gibt es auch ein Hydrocortison-Präparat mit verzögerter Wirkstofffreisetzung, dass extra für Menschen mit AGS entwickelt wurde. Dieses muss nur zweimal am Tag eingenommen werden – morgens direkt nach dem Aufstehen und abends vor dem Einschlafen.